
Case report: Two cases of Hb Liuzhou-Yufeng (HBA1:c.334G>T) found in Northern Thailand
Article Sidebar
Main Article Content
Abstract
Background: Hemoglobin (Hb) Liuzhou-Yufeng is an α-globin variant caused by a heterozygous HBA1 mutation (c.334G>T). However, its clinical significance is not well-defined. Moreover, it is hard to detect using standard capillary electrophoresis (CE) or high-performance liquid chromatography (HPLC) methods and often need molecular techniques like next-generation sequencing (NGS).
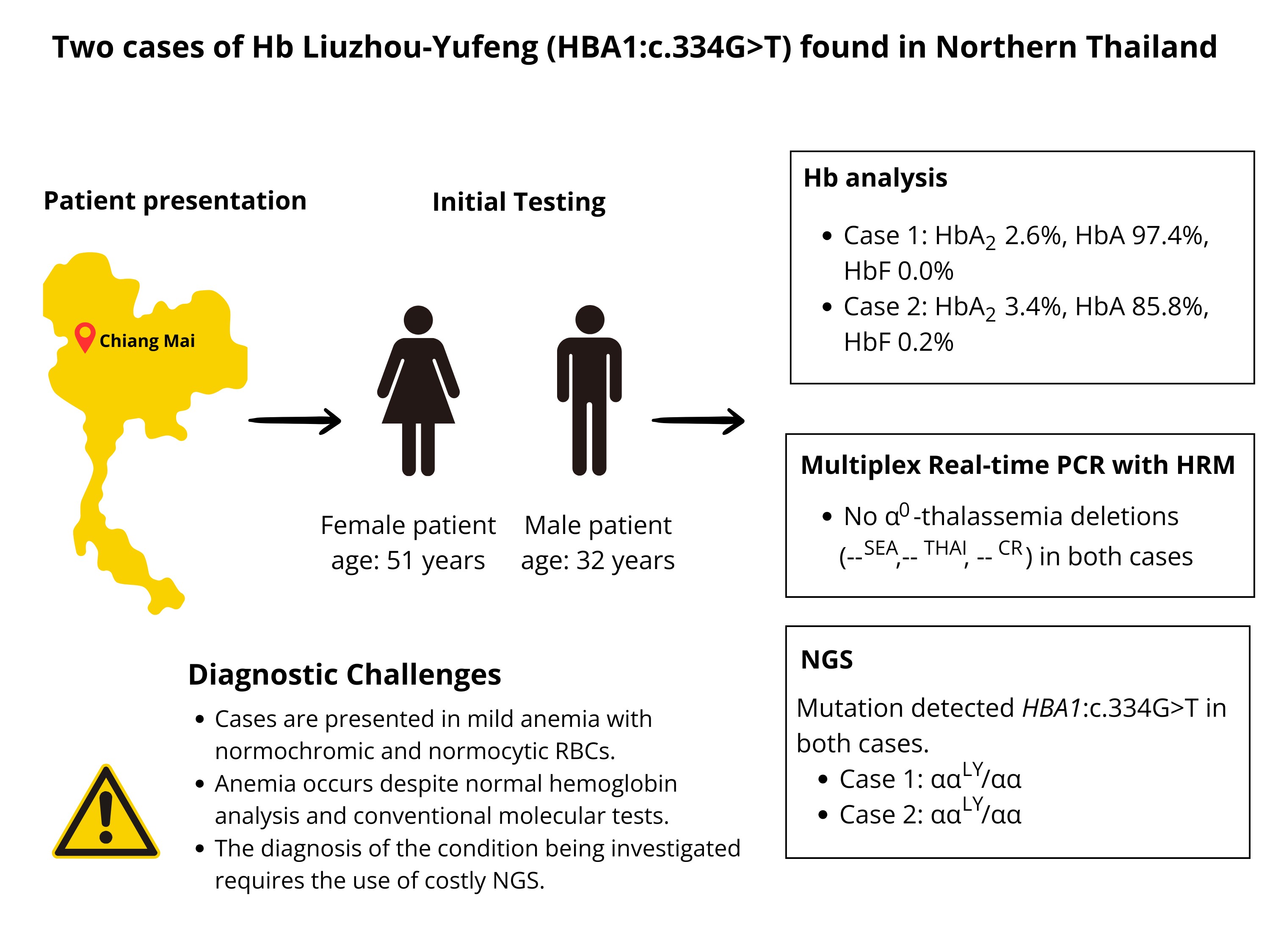
Objective: This study aims to describe genotype and hematological features of the first two cases with heterozygosity for Hb Liuzhou-Yufeng found in Northern Thailand and the diagnostic challenges faced.
Materials and methods: Two patients with anemia, a 51-year-old female and a 32-year-old male, underwent hematological evaluation, Hb analysis using CE for case 1 and HPLC for case 2, iron studies, and red cell morphology tests. The dichlorophenolindophenol (DCIP) and osmotic fragility (OF) tests were also conducted. The molecular work-up included multiplex real-time PCR for α-thalassemia deletions, followed by NGS.
Results: Both patients were confirmed by the NGS to carry Hb Liuzhou-Yufeng (ααLY/αα). Their total Hb levels were found within the ranges of mild anemia (10-12 g/dL). However, they had normocytic and normochromic red cells, and the morphology of the red blood cells appeared to be normal. In addition, the Hb analysis by HPLC and CE methods revealed normal peaks and levels of HbA, HbA2, and HbF.
Conclusion: Hb Liuzhou-Yufeng seems clinically harmless but is challenging to detect using conventional methods. This study highlights the importance of NGS in finding rare Hb variants. It improves diagnostic accuracy, genetic counseling, and personalized management of hemoglobinopathy.
Article Details

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Personal views expressed by the contributors in their articles are not necessarily those of the Journal of Associated Medical Sciences, Faculty of Associated Medical Sciences, Chiang Mai University.
References
Leckngam P. Thalassemia and hemoglobinopathies in Thailand: A systematic review. J Health Sci Altern Med. 2023; 5(3): 104-13. doi: 10.14456/jhsam.2023.24.
WHO. Regional desk review of haemoglobinopathies with an emphasis on thalassaemia and accessibility and availability of safe blood and blood products as per these patients’ requirement in South-East Asia under universal health coverage [Internet]. 2021.
Available from: http://apps.who.int/bookorders.
Jomoui W, Panyasai S, Sripornsawan P, Tepakhan W. Revisiting and updating molecular epidemiology of α-thalassemia mutations in Thailand using MLPA and new multiplex gap-PCR for nine α-thalassemia deletion. Sci Rep. 2023; 13(1): 9850. doi: 10.1038/s41598-023-36840-8.
Musallam KM, Cappellini MD, Coates TD, Kuo KHM, Al-Samkari H, Sheth S, et al. Αlpha-thalassemia: A practical overview. Blood Rev. 2024; 64: 101165. doi: 10.1016/j.blre.2023.101165.
Paiboonsukwong K, Jopang Y, Winichagoon P, Fucharoen S. Thalassemia in Thailand. Hemoglobin. 2022; 46(1): 53-7. doi:10.1080/03630269.2022.2025824.
Xu A, Chen W, Xie W, Ji L. Identification of a new hemoglobin variant Hb Liuzhou [HBA1:C.182A→G] by MALDI-TOF mass spectrometry during HbA1c measurement. Scand J Clin Lab Invest. 2020; 80(6): 479-83. doi: 10.1080/00365513.2020.1783698.
Ruengdit C, Punyamung M, Intasai N, Pornprasert S. Single-tube multiplex real-time PCR with EvaGreen and high-resolution melting analysis for diagnosis of α0-thalassemia--SEA,--THAI, and--CR type deletions. PLoS One. 2023; 18(11): e0293838. doi: 10.1371/journal.pone.0293838.
Fucharoen S, Fucharoen G, Sanchaisuriya K, Pengjam Y. Molecular analysis of a Thai β-thalassaemia heterozygote with normal
haemoglobin A2 level: implication for population screening. Ann Clin Biochem. 2002; 39(1): 44-9. doi: 10.1258/0004563021901720.
Traeger-Synodinos J, Vrettou C, Sofocleous C, Zurlo M, Finotti A, Gambari R. Impact of α-globin gene expression and α-globin modifiers on the phenotype of β-thalassemia and other hemoglobinopathies: Implications for patient management. Int J Mol Sci. 2024; 25(6): 3400. doi: 10.3390/ijms25063400.
Vijian D, Wan Ab Rahman WS, Ponnuraj KT, Zulkafli Z, Bahar R, Yasin N, et al. Gene mutation spectrum among alpha-Thalassaemia patients in Northeast Peninsular Malaysia. Diagnostics. 2023; 13(5): 894. doi: 10.3390/diagnostics13050894.
Knappenberger JA, Kuriakose SA, Vu BC, Nothnagel HJ, Vuletich DA, Lecomte JTJ. Proximal influences in two-on-two globins: Effect of the Ala69Ser replacement on synechocystis sp. PCC 6803 hemoglobin. Biochemistry. 2006; 45(38): 11401-13. doi: 10.1021/bi060691x.
Narayanan S, Mathew B, Srinivasu BY, Bhat V, Ross C, Mandal AK. Effect of point mutation on structure–function correlation of hemoglobin variants, HbE and HbD Punjab. Amino Acids. 2020; 52(6-7): 893-904. doi: 10.1007/s00726-020-02858-9.
Pornprasert S, Ruengdit C, Punyamung M, Sripichai O. Alpha-Thalassemia caused by ααIVSI-1(AGGT> AGAT) (HBA1: c.95 + 1G > A) Mutation and its combinations with other forms of thalassemia or hemoglobinopathy in Northern Thailand. Indian J Hematol Blood Transfus. 2025; 41(3): 674-9. doi: 10.1007/s12288-024-01895-8.
Panyasai S, Chantanaskulwong P, Permsripong N, Mokmued T. Interactions of electrophoretically silent hemoglobin Hekinan II [HBA1:c.84G>T] with various forms of α-thalassemias and other hemoglobinopathies: novel insights into the molecular and hematological characteristics and genetic origins. Libyan J Med. 2024; 19(1): 2406620. doi: 10.1080/19932820.2024.2406620.
