
Case Report: Two cases of Hb Malay (HBB: c.59A>G) found in Northern Thailand
Article Sidebar
Main Article Content
Abstract
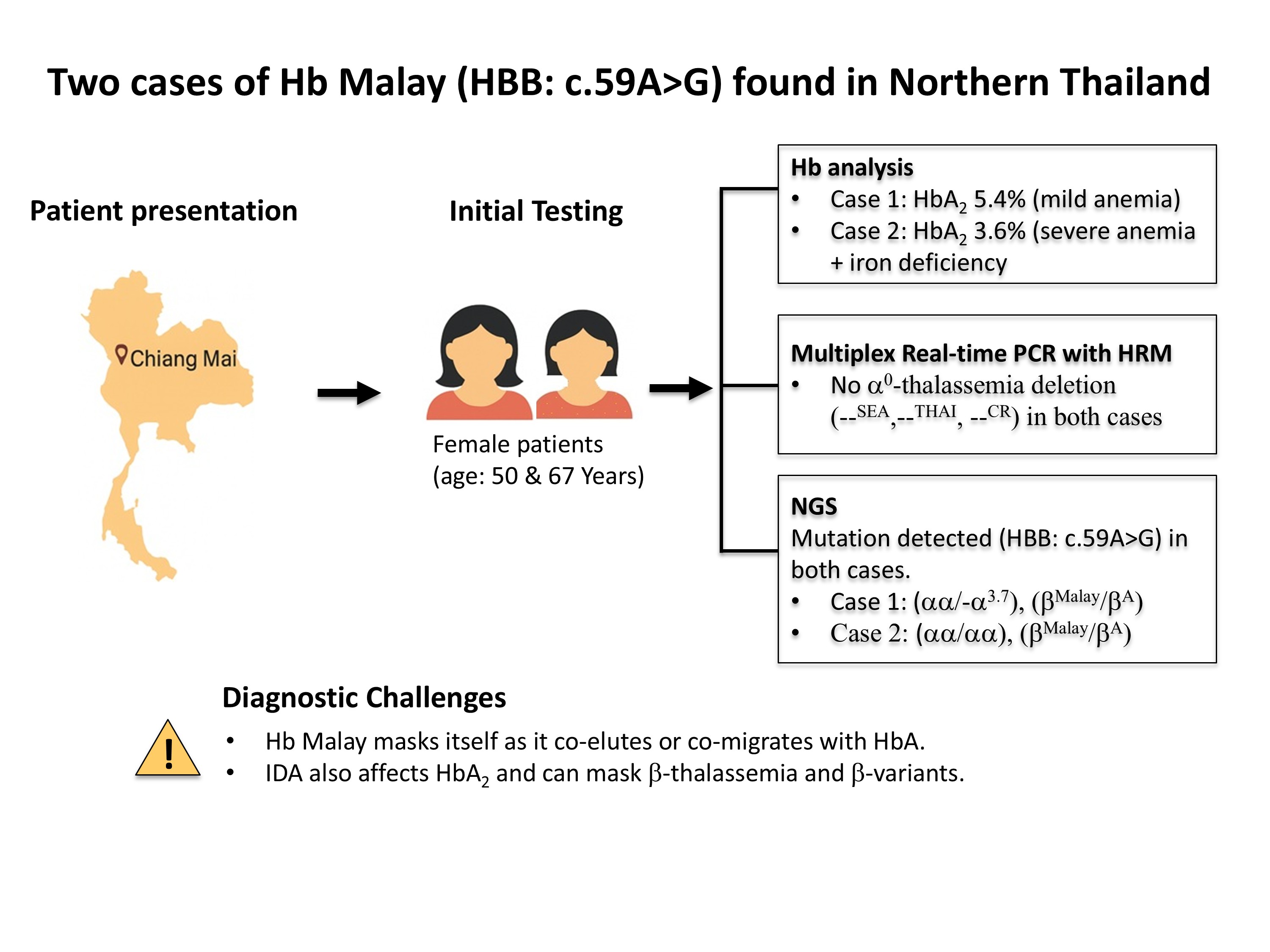
Background: Hemoglobin (Hb) Malay is a common β-hemoglobinopathy in Malaysia resulting from an AAC to AGC mutation at codon 19, which produces an abnormal β-globin chain and manifests as a β+-thalassemia phenotype characterized by mild anemia and increased HbA2 levels. Although Hb Malay is commonly prevalent in Southern Thailand, there have been few reported cases in Northern Thailand.
Objectives: This study aims to report two cases of Hb Malay detected in Chiang Mai, Northern Thailand, highlighting the diagnostic complexities. Materials and methods: Two female patients, aged 50 and 67, presenting with anemia were investigated. Initial hematological profiles, Hb analysis by HPLC (Case 1) and CE (Case 2), and iron studies were performed. Due to hypochromicmicrocytic
anemia and elevated HbA2 levels in both cases, genomic DNA was extracted. Multiplex real-time PCR with HRM analysis was performed to detect common α0-thalassemia deletions. Further genetic analysis was conducted using next-generation sequencing (NGS) targeting HBA1, HBA2, and HBB genes.
Results: Both cases were identified to carry the Hb Malay variant. Case 1, a 50- year-old female with mild anemia, was diagnosed with double heterozygosity for Hb Malay (βMalay/βA) and α+-thalassemia (-α3.7/αα). Case 2, a 67-year-old female with severe anemia and iron deficiency, was diagnosed with heterozygosity for Hb Malay (βMalay/βA).
Conclusion: The diagnosis of Hb Malay can be complicated especially when it coexists with other thalassemia traits or iron deficiency. Therefore, better understanding of the hematological and clinical characteristics, as well as the laboratory detection of this hemoglobin variant, would be beneficial for genetic counseling, particularly in areas with a high prevalence of thalassemia, hemoglobinopathy, and iron deficiency such as Northern Thailand.
Article Details

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Personal views expressed by the contributors in their articles are not necessarily those of the Journal of Associated Medical Sciences, Faculty of Associated Medical Sciences, Chiang Mai University.
References
Yang KG, Kutlar F, George E, Wilson JB, Kutlar A, Stoming TA, et al. Molecular characterization of β-globin gene mutations in Malay patients with Hb E-β-thalassaemia and thalassaemia major. Br J Haematol. 1989;72(1):73-80. doi: 10.1111/j.1365-2141.1989.tb07655.x.
Leckngam P. Thalassemia and Hemoglobinopathies in Thailand: A Systematic Review. j health sci altern med [internet]. 2023;5(03):104-13. [cited 2025 Jun. 29] available from: https://he01.tci-thaijo.org/index.php/jhealthscialternmed/article/view/262338.
Paiboonsukwong K, Jopang Y, Winichagoon P, Fucharoen S. Thalassemia in Thailand. Hemoglobin. 2022;46(1):53-7. doi: 10.1080/03630269.2022.2025824.
George E, Huisman TH, Yang KG, Kutlari F, Wilson JB, Kutlar A, et al. First observation of haemoglobin Malay alpha 2B2 26 (B1) Asn----Ser--a case report. Med J Malaysia. 1989;44(3):259-62.
Yamsri S, Singha K, Prajantasen T, Taweenan W, Fucharoen G, Sanchaisuriya K, et al. A large cohort of β+-thalassemia in Thailand: Molecular, hematological and diagnostic considerations. Blood Cells Mol. Diseases. 2015;54(2):164-9. https://doi.org/10.1016/j.bcmd.2014.11.008.
Bahar R, Shahida S N, Hassan mn, Marini R, mohd noor nh, Shafini M Y, et al. The Diagnosis of Beta Thalassemia with Borderline HbA2 Level among Kelantan Population. J Blood Disord Transfus. 2017;08(05). doi: 10.4172/2155-9864.1000396.
Tepakhan W, Kanjanaopas S, Sreworadechpisal K, Penglong T, Sripornsawan P, Wangchauy C, et al. Molecular epidemiology and hematological profiles of hemoglobin variants in southern Thailand. Sci Rep. 2024;14(1):9255. doi: 10.1038/s41598-024-59987-4.
Ruengdit C, Punyamung M, Intasai N, Pornprasert S. Single-tube multiplex real-time PCR with EvaGreen and high-resolution melting analysis for diagnosis of alpha0-thalassemia--SEA,--THAI, and--CR type deletions. PLoS One. 2023;18(11):e0293838. doi: 10.1371/journal.pone.0293838.
Li R, Shen X, Chen H, Peng D, wu R, Sun HY. Developmental validation of the MGIEasy Signature Identification Library Prep Kit, an all-in-one multiplex system for forensic applications. Int J Legal Med. 2021;135(3):739-53. doi: 10.1007/s00414-021-02507-0.
Pornprasert S, Thichak S, Kongthai K, Wangchauy C. Comparison of HbA2, E, F and Red Cell Parameters in Homozygous HbE With and Without α0-Thalassemia Trait. Lab Med. 2017;49(2):118-22. doi: 10.1093/labmed/lmx083.
Fucharoen G, Trithipsombat J, Sirithawee S, Yamsri S, Changtrakul Y, Sanchaisuriya K, et al. Molecular and hematological profiles of hemoglobin EE disease with different forms of alpha-thalassemia. Ann Hematol. 2006;85(7):450-4. doi: 10.1007/s00277-006-0093-5.
Winichagoon P, Fucharoen S, Chen P, Wasi P. Genetic Factors Affecting Clinical Severity in β-Thalassemia Syndromes. J Pediatr Hematol Oncol. 2000;22(6):573-80. doi: 10.1097/00043426-200011000-00026.
Passarello C, Giambona A, Cannata M, Vinciguerra M, Renda D, Maggio A. Iron deficiency does not compromise the diagnosis of high HbA(2) β thalassemia trait. Haematologica. 2012;97(3):472-3. doi: 10.3324/haematol.2011.052357.
Asri AS, Samsuddin MH, Jalil N, Mohamad Tahir N, Hashim H, Azma RZ, et al. Characterization of Hemoglobin Malay Phenotypes in Tertiary Hospitals. Hemoglobin. 2024;48(3):153-60. doi: 10.1080/03630269.2024.2380873.
Verma S, Gupta R, Kudesia M, Mathur A, Krishan G, Singh S. Coexisting iron deficiency anemia and Beta thalassemia trait: effect of iron therapy on red cell parameters and hemoglobin subtypes. ISRN Hematol. 2014;2014:293216. doi: 10.1155/2014/293216.
