
Non-transfusion dependent HbE/βO-thalassemia as the results of co-existent SEA-αO thalassemia, Hb Constant Spring, and XmnI-Gγ site: Thai family studies
Article Sidebar
Main Article Content
Abstract
Background: Four university students of northern Thai descent were found to be HbE/βO-thalassemia. However, they all had a mild form of this disease, categorized as Non-Transfusion Dependent Thalassemia.
Objectives: To analyze involvement of types of β-globin mutations, α-thalassemia, and XmnI-Gγ site in mild clinical symptoms observed in four Thai non-transfusion dependent HbE/βO-thalassemia cases.
Materials and methods: EDTA blood samples were collected from the patients and their family members after signing the informed consent. Automated complete blood count with blood smear examination, hemoglobin typing, molecular analysis for α and β-globin mutations, β-globin gene haplotypes, and XmnI-Gγ site were performed on all blood samples. In addition, nucleotide sequencing of β-globin gene and globin chain separation were performed for patient#3 and their parents.
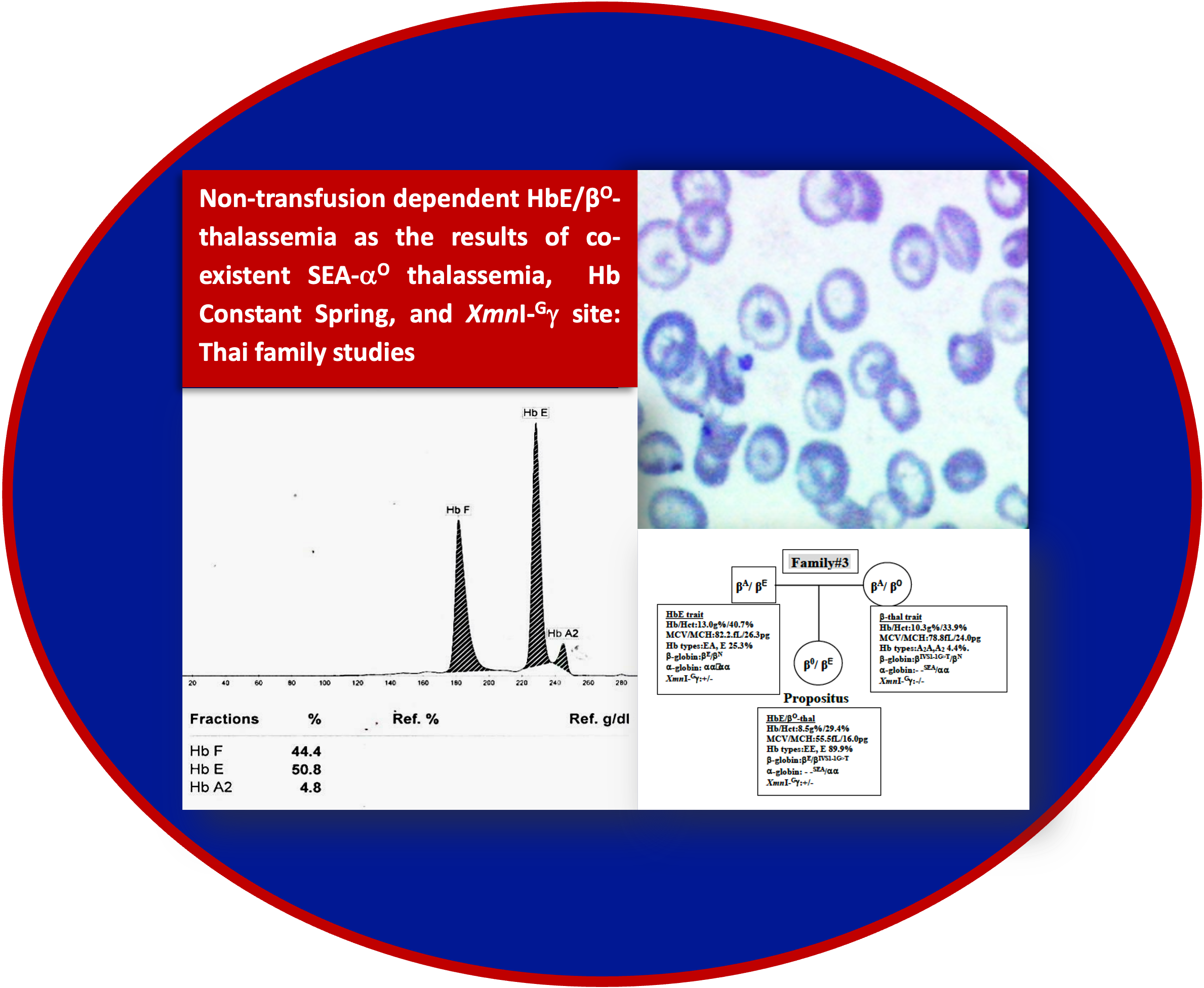
Results: The first three patients had hemoglobin levels ranging 8.5-11.2 g/dL, while the fourth patient had hemoglobin level of 6.7 g/dL. The first and fourth patients were compound heterozygote for βE (HBB:c.79G>A) and β17 (HBB:c.52A>T) alleles with typical hemoglobin pattern of EF. The second patient was compound heterozygote for βE and β41/42 (HBB:c.126_129delCTTT) alleles also with typical hemoglobin pattern of EF. The third patient was compound heterozygote of βE and βIVS1-1(HBB:c.92+1G>T), however, with atypical hemoglobin pattern of EE. Family analysis found co-inheritance of Hb Constant Spring (HBA2:c.427T>C) and the XmnI-Gγ site (T at rs7482144) in the first two patients, of SEA-αO thalassemia (NG_000006.1:g.26264_45564del19301) and XmnI-Gγ site in the third patient, and of only XmnI-Gγ site in the fourth patient.
Conclusion: These family studies proved the fact that co-existence of SEA-αO thalassemia and Hb Constant Spring in HbE/βO-thalassemia could lead to mild clinical severity. Minimal effect of XmnI-Gγ site on clinical symptoms of this disease was emphasized. This information should be useful in prenatal diagnosis of HbE/β-thalassemia.
Article Details

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Personal views expressed by the contributors in their articles are not necessarily those of the Journal of Associated Medical Sciences, Faculty of Associated Medical Sciences, Chiang Mai University.
References
Weatherall DJ, Clegg JB. The Thalassaemia Syndromes. 4th Ed. Oxford: Blackwell Scientific, 2001. doi:10.1002/ 9780470696705
Fucharoen S, Weatherall DJ (2012) The hemoglobin E thalassemias. Cold Spring Harb Perspect Med. 2012; 2(8): 1-15. doi: 10.1101/cshperspect.a011734.
Thein SL. Genetic modifiers of β-thalassemia. Haematologica 2005; 90(5): 649-60. PMID: 15921380
Musallam KM, Cappellini MD, Viprakasit V, et al. Revisiting the non-transfusion-dependent (NTDT) vs. transfusion-dependent (TDT) thalassemia classification 10 years later. Am J Hematol. 2021; 96(2): E54-E56. doi: 10.1002/ajh.26056.
Tatu T, Kiewkarnkha T, Khuntarak S, et al. Screening for co-existence of α-thalassemia in β-thalassemia and in HbE heterozygotes via an enzyme-linked immunosorbent assay for Hb Bart’s and embryonic ζ-globin chain. Int J Hematol. 2012; 95(4): 386-93. doi: 10.1007/s12185-012-1039-4.
Srisuwan W, Tatu T. Diagnosis of thalassemia carriers commonly found in northern Thailand via a combination of MCV or MCH and PCR-based methods. Bull Chiang Mai Assoc Med Sci. 2013; 46(1): 22-32.
Tatu T, Sritong W, Sa-Nguansermsri T. The associations of SEA-α thalassemia 1, XmnI-Gγ polymorphism and β-globin gene mutations with the clinical severity of β-thalassemia syndrome in northern Thailand. J Med Assoc Thai. 2014; 97(3): 300-7. PMID: 25123009
Kerdpoo S, Limweeraprajak E, Tatu T. Effect of Swisstype heterocellular HPFH from XmnI-Gγ and HBBP1 polymorphisms on HbF, HbE, MCV and MCH levels in Thai HbE carriers. Int J Hematol. 2014; 99(3): 338-44. doi: 10.1007/s12185-014-1516-z.
Dos Santos Silva W,de Nazare Klautau-Guimaraes M, Grisolia CK. β-globin haplotypes in normal and hemoglobinopathic individuals from Reconcavo Baiano, State of Bahia, Brazil. Genet Mol Biol. 2010; 33(3): 411-7. doi: 10.1590/S1415-47572010005000042.
Sutton M, Bouhassira EE, Nagel RL (1989) Polymerase chain reaction amplification applied to the determination of β-like globin gene cluster haplotypes. Am J Hematol. 1989; 32(1): 66-9. doi: 10.1002/ajh.2830320113.
Sripichai O, Makarasara W, Munkongdee T, et al. A scoring system for the classification of β-thalassemia/ Hb E disease severity. Am J Hematol. 2008; 83(6): 482-4. doi: 10.1002/ajh.21130.
Galanello R, Cao A. Relationship between genotype and phenotype. Thalassemia intermedia. Ann NY Acad Sci. 1998; 850: 325-33. doi: 10.1111/j.1749- 6632.1998.tb10489.x.
Cao A, Galanello R, Rosatelli MC. Genotype-phenotype correlations in β-thalassemias. Blood Rev. 1994; 8(1): 1-12. doi: 10.1016/0268-960x(94)90002-7.
Fucharoen S, Winichagoon P. Clinical and hematologic aspects of hemoglobin E beta-thalassemia. Curr Opin Hematol. 2000; 7(2): 106-12. doi: 10.1097/00062752- 200003000-00006.
Traivaree C, Monsereenusorn C, Rujkijyanont P, et al. Genotype-phenotype correlation among β-thalassemia and β-thalassemia/HbE disease in Thai children: predictable clinical spectrum using genotypic analysis. J Blood Med. 2018; 9: 35-41. doi: 10.2147/JBM.S159295. eCollection 2018.
Viprakasit V, Ekwattanakit S. Clinical Classification, Screening and Diagnosis for Thalassemia. Hematol Oncol Clin North Am. 2018; 32(2): 193-211. doi: 10.1016/j.hoc.2017.11.006.
Laboratories of Computer Science & Engineering and Biochemistry & Molecular Biology at the Pennsylvania State University. Globin Gene Server. 1999. https://globin.bx.psu.edu/. Accessed 25 Sept 2021.
Laosombat V, Wongchanchailert M, Sattayasevana B, et al. Clinical and hematologic features of βO-thalassemia (frameshift 41/42 mutation) in Thai patients. Haematologica. 2001; 86(2): 138-41. PMID: 11224481
Laosombat V, Wongchanchailert M, Sattayasevana B, et al (2001) Clinical and hematological features of codon 17, A-T mutation of β-thalassemia in Thai patients. Eur J Haematol. 2001; 66(2): 126-9. D doi: 10.1034/j.1600-0609.2001.00305.x.
Hunt DM, Higgs DR, Winichagoon, P, et al. Haemoglobin
Constant Spring has an unstable α chain messenger RNA. Br J Haematol. 1982; 51(3): 405-13. doi: 10.111 1/j.1365-2141.1982.tb02796.x.
Waggoner SA, Liebhaber SA. Regulation of α-globin mRNA stability. Exp Biol Med. (Maywood) 2003; 228(4): 387-95. doi: 10.1177/153537020322800409.
Bernini LF, Harteveld CL (1998) Alpha-Thalassemia. In: Rodgers GP, editor. Bailliere’s Clinical Haematology, International Practice and Research, Sickle cell disease and Thalassaemia. London: Bailliere Tindall; 1998. pp. 53-90.
Charoenkwan P, Taweephon R, Sae-Tung R, et al. Molecular and clinical features of Hb H disease in northern Thailand. Hemoglobin. 2005; 29(2): 133-40. PMID: 15921165
Fucharoen S, Viprakasit V. Hb H disease: clinical course and disease modifiers. Hematology Am Soc Hematol Educ Program. 2009: 26-34. doi: 10.1182/ asheducation-2009.1.26.
Garner C, Tatu T, Reittie JE, et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood. 2000; 95(1): 342-6. PMID: 10607722
Thein SL, Craig JE. Genetics of Hb F/F cell variance in adults and heterocellular hereditary persistence of fetal hemoglobin. Hemoglobin. 1998; 22(5-6): 401-14. doi: 10.3109/03630269809071538.
Thein SL, Menzel S. Discovering the genetics underlying foetal haemoglobin production in adults. Br J Haematol. 2009; 145(4): 455-67. doi:10.1111/j.1365-2141.2009. 07650.x.
Thein SL, Menzel S, Lathrop M, et al. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet. 2009; 18(R2): R216-23. doi: 10.1093/hmg/ddp401.
Alaoui-Ismaili FZ, Laghmich A, Ghailani-Nourouti N, et al. XmnI polymorphism in sickle cell disease in north Morocco. Hemoglobin. 2020; 44(3): 190-4. doi: 10.1080/03630269.2020.1772284.
Laosombat V, Wongchanchailert M, Sattayasevana B, et al. Clinical and hematological features of β+- thalassemia (IVS-1 nt 5, G-C mutation) in Thai patients. Eur J Haematol 2001; 67(2): 100-4. doi: 10.1034/j.1600-0609.2001.t01-1-00431.x.
Winichagoon P, Fucharoen S, Chen P, et al. Genetic factors affecting clinical severity in beta-thalassemia syndromes. J Pediatr Hematol Oncol. 2000; 22(6): 573-80. doi: 10.1097/00043426-200011000-00026.
Charoenkwan P, Teerachaimahit P, Sanguansermsri T. The correlation of α-globin gene mutations and the XmnI polymorphism with clinical severity of Hb E/β-thalassemia. Hemoglobin. 2014; 38(5): 335-8. doi: 10.3109/03630269.2014.952744.
Winichagoon P, Fucharoen S, Wilairat P, Chihara K, Fukumaki Y. Role of alternatively spliced βE-globin mRNA on clinical severity of β-thalassemia/ hemoglobin E disease. Southeast Asian J Trop Med Public Health. 1995;26(Suppl 1):241-5. PMID: 8629114
Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010; 5: 13. doi: 10.1186/1750-1172-5-13.
Higgs DR, Weatherall DJ. The Alpha Thalassaemias. Cell Mol Life Sci. 2009; 66(7): 1154-62. doi: 10.1007/ s00018-008-8529-9.
Sripichai O, Munkongdee T, Kumkhaek C, et al. Coinheritance of the different copy numbers of α-globin gene modifies severity of β-thalassemia/ Hb E disease. Ann Hematol. 2008; 87(5): 375-9. doi: 10.1007/s00277-007-0407-2.
Sharma V, Kumar B, Kumar G, et al. Alpha globin gene numbers: an important modifier of HbE/β thalassemia. Hematology. 2009; 14(5): 297-300. doi: 10.1179/102453309X446126.
